
Huntington’s Disease – Symptoms, Causes, and Treatment Option
Huntington’s disease is a genetic brain disorder that causes progressive changes in movement, thinking, and behavior. Early signs often include uncontrolled movements, mood swings, and difficulty concentrating, which worsen over time and impact daily life.
Many people with Huntington’s disease find support and improved function with physiotherapy for Huntington’s disease, where tailored exercises help maintain mobility, coordination, and strength. Massage for Huntington’s disease can ease muscle stiffness, reduce tension, and promote relaxation for greater comfort. Chiropractic care for Huntington’s disease may also support spinal alignment and joint mobility, helping reduce strain on the body. Working with a psychotherapist for Huntington’s disease provides emotional support, coping strategies, and guidance for managing the challenges of living with a progressive condition. Together, these services create a compassionate, holistic approach to improving quality of life.
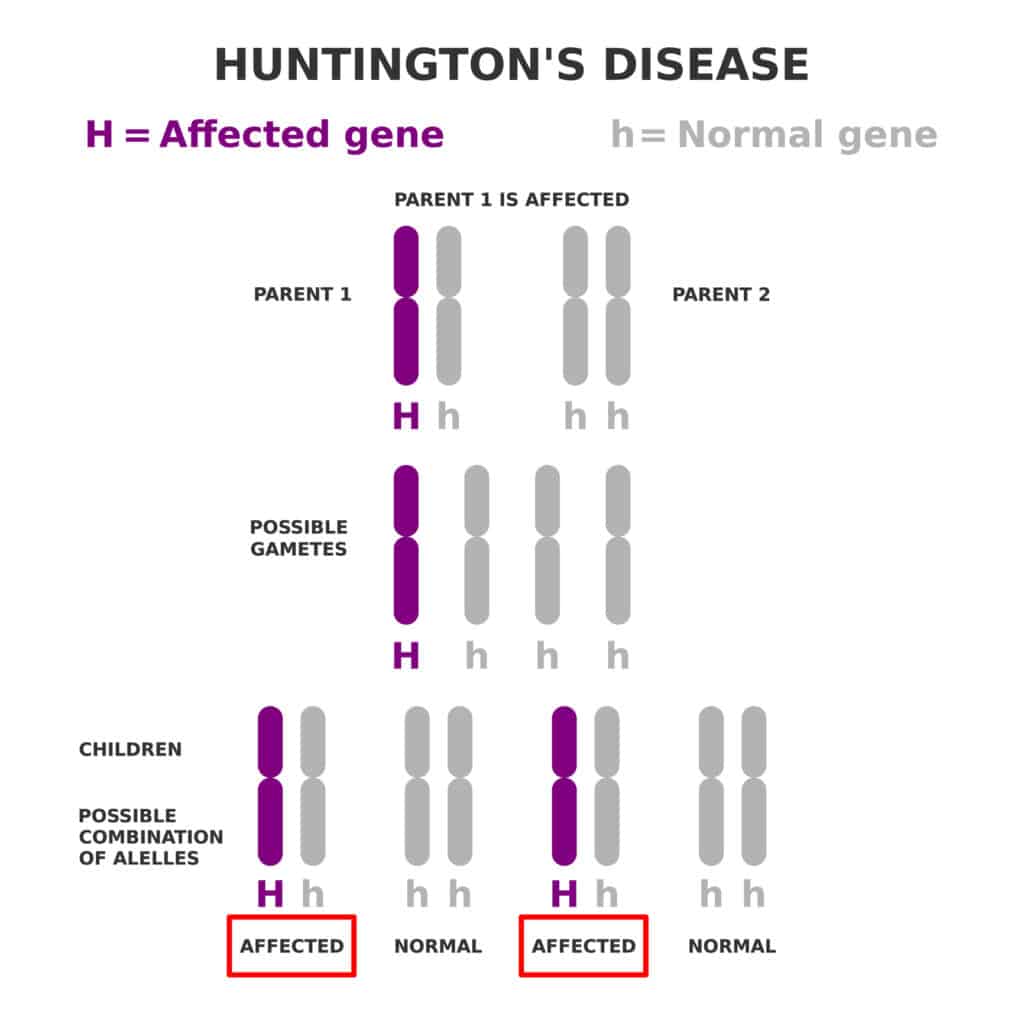
Overview of Huntington’s Disease
Huntington’s disease (HD) is a rare, inherited condition that gradually damages nerve cells in the brain, leading to movement, cognitive, and psychiatric symptoms. It is caused by a mutation in the HTT gene, which leads to abnormal production of a protein called huntingtin. This faulty protein builds up in brain cells, disrupting normal function and eventually causing cell death.
How common is Huntington’s disease?
Huntington’s disease affects approximately 3 to 7 people per 100,000 worldwide, with higher prevalence in populations of European descent. Because it is an autosomal dominant genetic disorder, a child of an affected parent has a 50% chance of inheriting the mutation. Symptoms usually begin between ages 30 and 50, though childhood (juvenile HD) and late-onset forms also exist.
Daily life impact
Huntington’s disease touches nearly every part of life:
- Work: Concentration difficulties, slower thinking, and involuntary movements often make it hard to maintain employment.
- Sport and activity: Balance problems and coordination issues increase risk of falls and injuries.
- Sleep: Sleep disturbances are common, leading to fatigue and worsening cognitive symptoms.
- Relationships: Personality changes, irritability, or depression can strain family and social bonds.
- Mental health: Anxiety, depression, and loss of independence are frequent challenges, both for individuals and caregivers.
How is Huntington’s disease different from similar conditions?
- Parkinson’s disease: Both affect movement, but Huntington’s typically starts earlier and involves more involuntary “dance-like” movements (chorea).
- Alzheimer’s disease: Both impair cognition, but Huntington’s includes pronounced motor symptoms and a clear genetic cause.
- ALS (Lou Gehrig’s disease): ALS causes muscle weakness and paralysis but usually spares thinking, while Huntington’s impairs both movement and cognition.
Reference:
Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. (2012). The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Movement Disorders, 27(9), 1083–1091. https://doi.org/10.1002/mds.25075
What are the main symptoms of Huntington’s Disease?
The symptoms of Huntington’s disease are a mix of motor (movement), cognitive (thinking), and psychiatric (emotional) changes. They progress gradually and vary from person to person.
Motor symptoms
- Chorea: Involuntary, jerky, dance-like movements.
- Poor coordination: Clumsiness, frequent falls, or difficulty with fine motor skills.
- Slurred speech and swallowing problems: These progress over time and may affect nutrition and communication.
- Muscle rigidity and dystonia: Stiff or abnormal postures, especially in advanced stages.
Cognitive symptoms
- Difficulty concentrating: Trouble focusing on tasks or conversations.
- Memory problems: Forgetfulness, especially with new information.
- Poor judgment: Difficulty making decisions or planning ahead.
- Slowed processing: Taking longer to understand or respond.
Psychiatric symptoms
- Depression: Common and may precede motor symptoms.
- Anxiety and irritability: Emotional volatility that can affect relationships.
- Obsessive-compulsive tendencies: Repetitive thoughts or actions.
- Psychosis (rare): Hallucinations or delusions in some cases.
Severity spectrum and red flag signs
- Early stage: Subtle personality changes, mild clumsiness, or small memory lapses.
- Middle stage: Noticeable movement difficulties, significant mood swings, and impaired work ability.
- Late stage: Severe motor disability, difficulty speaking or swallowing, full dependence on caregivers.
Red flag signs include new unexplained involuntary movements, personality changes, or a family history of Huntington’s disease. Genetic counseling and testing are strongly recommended if you have a parent with the condition.
Daily impacts of symptoms
- Work: Cognitive decline and fatigue can make it difficult to sustain professional roles.
- Sport and leisure: Participation declines as coordination worsens.
- Sleep: Sleep-wake cycles often become disrupted, worsening daytime function.
- Relationships: Family members often face emotional and caregiving burdens.
- Mental health: The emotional toll of knowing the disease is progressive can affect both patients and loved ones.
Reference:
Ross CA, Aylward EH, Wild EJ, et al. (2014). Huntington disease: natural history, biomarkers and prospects for therapeutics. Nature Reviews Neurology, 10(4), 204–216. https://doi.org/10.1038/nrneurol.2014.24
Causes and Risk Factors for Huntington’s Disease
Huntington’s disease is caused by a single genetic mutation in the HTT gene, leading to abnormal expansion of CAG repeats. This results in a toxic huntingtin protein that damages brain cells, especially in regions controlling movement, mood, and cognition.
Genetic cause
- Autosomal dominant inheritance: A parent with Huntington’s disease has a 50% chance of passing the gene to each child.
- CAG repeat length: Normal ranges are fewer than 36 repeats. Individuals with 40 or more repeats will almost always develop the disease, while those with 36–39 may or may not show symptoms.
- Anticipation effect: In some families, the number of repeats increases in the next generation, leading to earlier onset.
Risk factors
- Family history: The only known risk factor. Having an affected parent is the strongest predictor.
- Age: Symptoms usually appear between 30–50 years old, but juvenile forms can appear before age 20.
- Sex: Men and women are equally affected.
- Ethnicity: Higher prevalence in individuals of European ancestry; lower in Asian and African populations.
Lifestyle and environmental modifiers
While lifestyle doesn’t cause Huntington’s disease, some factors may influence symptom severity or progression:
- Stress and poor sleep may worsen mood and cognitive symptoms.
- Diet and exercise may support brain health, though they cannot stop the disease.
- Mental health support can reduce the burden of depression and anxiety associated with the disease.
How is Huntington’s disease different from other genetic disorders?
Unlike many genetic conditions, Huntington’s disease:
- Has a clear predictive genetic test that can confirm diagnosis before symptoms.
- Has 100% penetrance when the mutation exceeds 40 CAG repeats, meaning all carriers will eventually develop symptoms.
- Is progressive and incurable, though symptom management can improve quality of life.
Reference:
Walker FO. (2007). Huntington’s disease. The Lancet, 369(9557), 218–228. https://doi.org/10.1016/S0140-6736(07)60111-1
Diagnosis, Recovery, and Management of Huntington’s Disease
Huntington’s disease (HD) is a progressive, inherited brain disorder that requires specialized diagnosis, careful management, and long-term planning. While there is currently no cure, evidence-based strategies can slow symptom progression, improve daily life, and support both patients and families.
How is Huntington’s Disease Diagnosed?
Huntington’s disease is diagnosed through a combination of clinical evaluation, neurological examination, family history review, and genetic testing. Because its symptoms overlap with other neurological and psychiatric disorders, accurate diagnosis is critical.
How do doctors test for Huntington’s disease?
- Medical history and family background
- A healthcare provider will ask about your symptoms and whether anyone in your family has been diagnosed with HD.
- Since Huntington’s is autosomal dominant, family history is a strong predictor.
- Neurological exam
- Doctors look for motor symptoms such as chorea (involuntary movements), coordination problems, and balance issues.
- They also assess reflexes, eye movements, and speech clarity.
- Cognitive and psychiatric evaluation
- Memory, attention, problem-solving, and emotional regulation are assessed through standardized tests and interviews.
- Depression, anxiety, and irritability are considered alongside physical symptoms.
- Genetic testing
- A blood test checks for abnormal CAG repeat expansions in the HTT gene.
- ≥40 repeats almost always confirms HD; 36–39 repeats may cause symptoms later in life or not at all.
- Genetic counseling is essential before and after testing to support decision-making.
- Imaging tests (optional but supportive)
- MRI or CT scans may show brain changes (such as shrinkage in the caudate nucleus) before symptoms appear.
- Imaging helps rule out other neurological conditions.
Identifying the root cause of symptoms
Doctors must differentiate HD from conditions like Parkinson’s, Alzheimer’s, or psychiatric illnesses. For example:
- Parkinson’s disease causes stiffness and tremors, not chorea.
- Alzheimer’s affects memory early but does not cause involuntary movements.
- Psychiatric disorders may mimic mood symptoms but lack neurological signs.
Reference:
Ross CA, et al. (2014). Huntington disease: natural history, biomarkers, and prospects for therapeutics. Nature Reviews Neurology, 10(4), 204–216. https://doi.org/10.1038/nrneurol.2014.24
Recovery Timeline and Prognosis for Huntington’s Disease
Huntington’s disease is progressive, meaning symptoms worsen over time. While recovery in the sense of reversal isn’t possible, management can extend independence, reduce complications, and improve quality of life.
How long does Huntington’s disease take to progress?
- Early stage (0–5 years after onset): Mild motor symptoms, mood swings, and cognitive changes. Most people remain independent and may continue working.
- Middle stage (5–15 years): Worsening chorea, slurred speech, difficulty swallowing, and memory decline. Assistance with daily activities is often required.
- Late stage (15–25 years): Severe motor disability, inability to speak, full dependence on caregivers. Life expectancy is typically 15–20 years after onset.
Decision pathway (if/then logic)
- If symptoms are mild → lifestyle strategies, counseling, and early rehabilitation can preserve independence.
- If symptoms are moderate → medical therapy, physiotherapy, and speech therapy support daily function.
- If symptoms are severe → palliative care, assisted living, and caregiver support become essential.
Factors influencing long-term outcomes
- CAG repeat length: More repeats often mean earlier onset and faster progression.
- Lifestyle: Exercise, diet, and cognitive stimulation may help maintain function longer.
- Access to care: Early interventions and multidisciplinary support improve quality of life.
- Mental health: Treating depression and anxiety improves resilience.
Return-to-work, return-to-sport, and lifestyle outcomes
- Work: Many continue in their careers early on but may need modified duties or reduced hours as cognition declines.
- Sport and exercise: Low-impact activities like swimming, walking, or yoga remain beneficial but may need adaptation for balance and safety.
- Lifestyle: A strong support network and structured routines help maintain dignity and independence for as long as possible.
Reference:
Roos RA. (2010). Huntington’s disease: a clinical review. Orphanet Journal of Rare Diseases, 5, 40. https://doi.org/10.1186/1750-1172-5-40
How to Manage Huntington’s Disease
Management of Huntington’s disease focuses on treating symptoms, maintaining function, and supporting mental health. A combination of medical, therapeutic, and lifestyle approaches is usually recommended.
Medical management
- Medications for movement symptoms: Tetrabenazine or deutetrabenazine may reduce chorea. Antipsychotics and benzodiazepines can also help with involuntary movements.
- Psychiatric treatment: Antidepressants, mood stabilizers, or antipsychotics may be used for depression, anxiety, or irritability.
- Nutritional support: High-calorie diets may be needed since chorea burns extra energy. Swallowing therapy helps prevent malnutrition.
Rehabilitation and therapy
- Physiotherapy: Improves balance, posture, and mobility.
- Occupational therapy: Helps adapt home and work environments for safety.
- Speech therapy: Supports communication and swallowing.
- Cognitive training: Exercises that challenge memory and reasoning may help slow decline.
Step-by-step self-help & relief tips
- Stay active: Gentle, regular exercise helps maintain strength and mood.
- Eat nutrient-rich meals: Support brain and body health with balanced, high-energy foods.
- Protect your environment: Remove tripping hazards to reduce fall risks.
- Maintain routines: Predictable daily structure reduces stress and confusion.
- Engage socially: Support groups and family activities help preserve emotional well-being.
- Prioritize mental health: Seek counseling or therapy early for mood-related symptoms.
Therapy, coaching, and social supports
- Psychotherapy and coaching: Help patients and families cope with stress, grief, and planning for the future.
- Community support groups: Provide emotional relief and practical advice from others living with HD.
- Caregiver education: Prepares families for progressive care needs.
How is managing Huntington’s disease different from similar conditions?
Unlike Parkinson’s or Alzheimer’s, management of HD requires addressing all three symptom areas (movement, thinking, mood) simultaneously. Because it is inherited and predictable, genetic counseling and family planning are central aspects of care.
Reference:
Frank S. (2014). Treatment of Huntington’s disease. Neurotherapeutics, 11(1), 153–160.
Multidisciplinary Coordinated Care for Huntington’s Disease at CARESPACE
At CARESPACE, we recognize that Huntington’s disease affects not only movement but also thinking, emotions, and daily living. That’s why our approach is multidisciplinary and coordinated—bringing together professionals across physical therapy, mental health, nutrition, and lifestyle services to give you and your family the most comprehensive support possible.
How does CARESPACE treat Huntington’s Disease differently?
CARESPACE treats Huntington’s disease with a whole-person strategy, not just a single treatment. Instead of focusing only on medications or movement symptoms, we build integrated care plans that address physical health, mental wellness, and family support.
- A physiotherapist helps improve strength, balance, and posture, reducing fall risk and maintaining mobility.
- A speech-language therapist supports communication and swallowing challenges that develop as the disease progresses.
- A psychotherapist provides counseling for depression, anxiety, or irritability, which are common in Huntington’s.
- A dietitian helps design high-calorie, nutrient-rich meal plans to prevent weight loss caused by chorea (involuntary movements).
- A massage therapist reduces muscle stiffness and promotes relaxation.
- A fitness trainer or kinesiologist builds safe exercise routines to maintain independence for as long as possible.
This integrated model means you don’t have to coordinate care across multiple clinics—our team works together to guide your journey.
Why does a team approach help Huntington’s Disease care succeed?
Huntington’s disease impacts the brain, body, and emotions all at once—making a team approach essential. A single type of care can’t fully manage the complex symptoms, but coordinated disciplines can.
Acute phase (early symptoms)
- Physiotherapy builds mobility strategies while occupational therapy adapts your home and work environment.
- Psychotherapy supports coping with the emotional burden of diagnosis.
- Nutrition planning helps sustain energy and weight.
Subacute phase (progressive symptoms)
- Speech therapy provides tools for communication and swallowing safety.
- Massage therapy and acupuncture relieve muscle tension and discomfort.
- Coaching and counseling help families adjust roles and caregiving responsibilities.
Maintenance phase (advanced care)
- Palliative support focuses on comfort, dignity, and quality of life.
- Psychotherapy continues to support both patients and caregivers.
- Multidisciplinary input ensures transitions in care are smooth and supportive.
How does CARESPACE support both body and mind in Huntington’s Disease?
At CARESPACE, care for Huntington’s disease extends beyond physical therapy—it includes emotional, psychological, and social health. This dual focus is what makes our coordinated care unique.
- Psychotherapy: Provides safe spaces for individuals and families to process grief, uncertainty, and stress.
- Mental performance coaching: Helps clients set realistic goals and maintain motivation as symptoms progress.
- Nutrition support: Guides eating strategies to reduce fatigue, stabilize energy, and protect overall health.
- Sleep and stress management: Coaching and lifestyle interventions reduce insomnia and improve coping.
This balance of mind and body care allows you and your family to navigate the disease with greater resilience.
Why is coordinated care better than isolated treatment for Huntington’s Disease?
Coordinated care at CARESPACE delivers advantages that single-discipline approaches can’t provide. When neurologists, therapists, dietitians, and mental health professionals work in silos, patients often receive fragmented support. At CARESPACE, your team communicates, shares notes, and aligns goals—so your treatment plan is seamless and personalized.
Key benefits of CARESPACE’s multidisciplinary model:
- Personalized plans: Tailored to your stage of Huntington’s and your lifestyle needs.
- Faster progress: Addressing symptoms from multiple angles speeds up improvements in comfort and function.
- Lower stress: One team coordinates care, reducing the burden on you and your family.
- Better prevention: Early interventions reduce fall risk, malnutrition, and mental health crises.
- Whole-family support: Caregivers receive as much support as clients, ensuring no one faces the journey alone.
For example, someone with early Huntington’s symptoms may start with physiotherapy to improve balance, while also seeing a dietitian to build a calorie-rich meal plan and a psychotherapist to manage anxiety. Together, this team-based care slows decline, boosts independence, and provides peace of mind for families.
The CARESPACE Advantage in Huntington’s Disease Care
CARESPACE’s strength lies in coordinated, evidence-based, and compassionate care. By combining physiotherapy, psychotherapy, nutrition, naturopathic medicine, massage therapy, and fitness training, we help you manage symptoms while supporting your emotional and social well-being.
This is more than treatment—it’s a partnership in care that adapts with you, reduces complications, and improves quality of life at every stage of Huntington’s disease.
Related Conditions, FAQs, and Disclaimer for Huntington’s Disease
Huntington’s disease affects the brain, body, and emotions in complex ways. Because its symptoms overlap with other conditions, understanding related disorders, frequently asked questions, and key compliance information can help you and your family feel more informed and supported.
Related Conditions for Huntington’s Disease
Several conditions share symptoms with Huntington’s disease, making diagnosis and management more challenging. Recognizing overlaps is important to avoid misdiagnosis and ensure proper treatment.
- Parkinson’s disease: Both cause movement issues, but Parkinson’s often features tremors and stiffness, while Huntington’s is known for jerky, dance-like movements (chorea).
- Alzheimer’s disease: Memory loss occurs in both conditions, but Huntington’s includes psychiatric changes and movement problems early on.
- ALS (amyotrophic lateral sclerosis): Like Huntington’s, ALS affects muscles, but it primarily causes weakness and paralysis without cognitive decline.
- Obsessive-compulsive disorder (OCD) or depression: Psychiatric symptoms in Huntington’s can mimic standalone mental health disorders, but are linked to neurological changes.
- Juvenile Huntington’s disease vs. epilepsy: Seizures may appear in early-onset Huntington’s, which can resemble epilepsy.
For a full list of overlapping conditions, please see our Conditions List.
Looking for information on a different condition? Visit our full Conditions List.
FAQs About Huntington's Disease
There’s no single “fast fix” for Huntington’s disease, but symptom relief can be achieved with a combination of therapies. Medications may reduce involuntary movements, while physiotherapy and occupational therapy improve balance and daily function. Counseling and stress management can ease emotional strain.
Quick relief usually means treating the most disruptive symptom first—for example, addressing chorea with medication or supporting depression with therapy. Long-term care requires a tailored, multidisciplinary approach.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Huntington’s disease does not go away on its own because it is caused by a genetic mutation. Unlike temporary conditions, HD is progressive and lifelong. However, treatments can help manage symptoms, maintain independence longer, and improve quality of life.
Lifestyle strategies such as exercise, healthy eating, and stress reduction can support brain and body health, but they cannot reverse the disease. Early care planning is key.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
You should see a doctor if you notice unexplained involuntary movements, personality changes, or cognitive decline—especially if Huntington’s disease runs in your family.
Early diagnosis allows for better planning, symptom management, and family support. If you already know you carry the Huntington’s gene, regular check-ins with a neurologist are essential. Seek care urgently if you experience sudden weight loss, difficulty swallowing, or mental health crises.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Lifestyle changes can’t stop Huntington’s disease but can make daily life easier. Helpful strategies include:
- Staying active with safe, low-impact exercise like walking or swimming.
- Eating high-calorie, nutrient-dense foods to prevent weight loss from chorea.
- Maintaining regular sleep routines to improve mood and energy.
- Creating a safe home environment to reduce fall risks.
- Building strong social and emotional support networks.
These habits won’t alter disease progression but can significantly improve quality of life.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Huntington’s disease affects men and women equally because it is inherited in an autosomal dominant pattern. Both sexes have a 50% chance of inheriting the mutation if one parent carries it.
That said, men and women may experience differences in symptoms such as psychiatric changes, with some studies suggesting depression may be more common in women. Disease severity is influenced more by genetic factors (CAG repeat length) than by sex.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Medication is not always required in the earliest stages of Huntington’s disease, but it becomes helpful as symptoms progress. Drugs like tetrabenazine may reduce involuntary movements, while antidepressants or antipsychotics can support mood stability.
Even when medication is not yet needed, therapies such as physiotherapy, nutrition planning, and counseling provide meaningful benefits. A doctor can guide whether and when medications are appropriate.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
You cannot stop Huntington’s disease from progressing, but you can slow its impact with proactive care. Key strategies include:
- Starting physiotherapy early to maintain balance and strength.
- Eating a nutrient-rich diet to avoid malnutrition.
- Using mental health support to manage stress, depression, and anxiety.
- Engaging in mentally stimulating activities to support cognitive function.
Prevention in this context means reducing complications and maintaining independence for as long as possible.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Huntington’s disease is unique because it combines movement, cognitive, and psychiatric symptoms—and it has a clear genetic cause.
- Compared to Parkinson’s disease: Huntington’s has earlier psychiatric changes and jerky movements instead of tremors.
- Compared to Alzheimer’s disease: Huntington’s involves motor decline as well as memory loss.
- Compared to ALS: Huntington’s affects thinking and emotions, while ALS primarily affects muscle weakness and breathing.
This combination makes Huntington’s distinct and underscores the importance of genetic testing and multidisciplinary care.
This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s disease, consult a qualified health provider.
Articles on Huntington's Disease
For more information, you can view all Huntington’s disease articles on our resource hub.
Authorship & Disclaimer
Reviewed by: Ellen Layton, Nurse Practitioner
Last Updated: September 1st, 2025
Disclaimer: This content is for informational purposes only and is not a substitute for professional medical advice. If you think you may have Huntington’s Disease, consult a qualified health provider.